Chapter 4 Differential Expression Analysis
This chapter focuses on differential expression analysis for cross-sectional studies, identifying proteins that differ significantly between groups using linear models.
4.1 Overview
Differential expression analysis answers the question: “Which protein abundance levels are significantly associated with a specific condition or phenotype?”
The lmNULISAseq() function:
- Fits a linear model for each protein
- Tests associations between protein levels and your primary variables of interest (e.g., Disease vs. Control)
- Adjusts for covariates, ensuring that differences are attributed by the effect of interest
- Corrects for multiple testing (Benjamini-Hochberg (BH) FDR, Bonferroni correction)
See complete function documentation and additional options, use ?lmNULISAseq().
4.3 Model Results
The function returns a list with:
#> [1] "modelStats"#> Preview of `lmTest$modelStats` Results Table (rounded to 3 digits):4.3.1 Key Columns in Results
For each comparison (e.g., “disease_typeinflam” vs reference “normal”):
disease_typeinflam_coef: Effect size (log fold-change)disease_typeinflam_pval: Raw p-valuedisease_typeinflam_pval_FDR: FDR-adjusted p-valuedisease_typeinflam_pval_bonf: Bonferroni-adjusted p-valuetarget: Target name
4.4 Finding Significant Proteins
Focusing on the comparison between Inflammation vs Normal, adjusting for age and sex. We will identify proteins that are significantly differentially expressed with FDR-adjusted p value < 0.05.
4.4.1 Filter by FDR Threshold
# Define significance threshold
fdr_threshold <- 0.05
fc_threshold <- 0.5 # Fold-change threshold (log scale)
# Find significant proteins for Inflammation vs Normal
sig_inflam <- lmTest$modelStats %>%
filter(
disease_typeinflam_pval_FDR < fdr_threshold,
abs(disease_typeinflam_coef) > fc_threshold
) %>%
arrange(disease_typeinflam_pval_FDR)#> Significant proteins (Inflammation vs Normal): 944.4.2 Upregulated vs Downregulated
We can further identify significantly differentially expressed proteins that are either unregulated or down regulated in the inflammation group comparing to normal group.
# Upregulated in inflammation
upregulated <- sig_inflam %>%
filter(disease_typeinflam_coef > 0) %>%
arrange(desc(disease_typeinflam_coef))
# Downregulated in inflammation
downregulated <- sig_inflam %>%
filter(disease_typeinflam_coef < 0) %>%
arrange(disease_typeinflam_coef)
cat("Upregulated:", nrow(upregulated), "\n")#> Upregulated: 94#> Downregulated: 04.5 Volcano Plots
Volcano plots visualize differential expression results effectively. See complete function documentation and additional options, use ?volcanoPlot().
4.5.1 Inflammation vs Normal
volcanoPlot(
coefs = lmTest$modelStats$disease_typeinflam_coef,
p_vals = lmTest$modelStats$disease_typeinflam_pval_FDR,
target_labels = lmTest$modelStats$target,
title = "Inflammation vs Normal"
)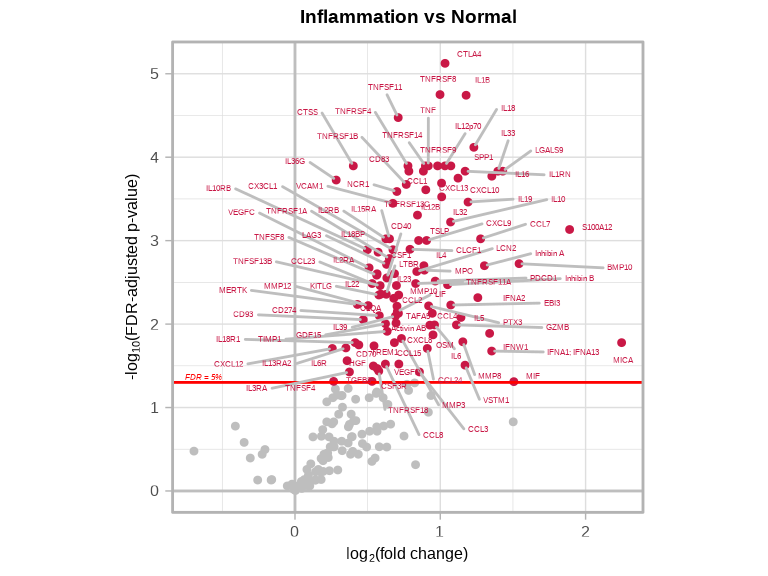
4.5.2 Understanding Volcano Plots
- X-axis: Effect size (log fold-change)
- Y-axis: Statistical significance (-log10 p-value)
- Significant targets: Points in upper left/right corners
- Upregulated targets: Right side/ Red (positive fold-change)
- Downregulated targets: Left side/ Blue (negative fold-change)
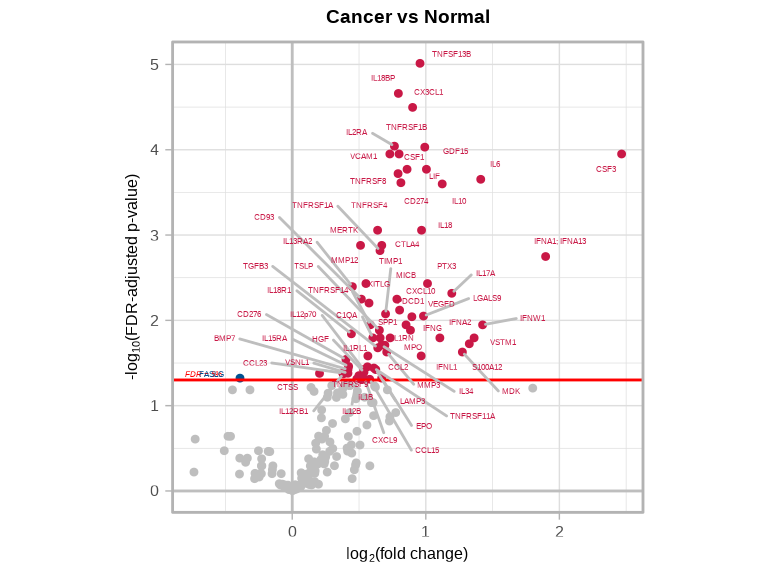
4.5.3 Cancer vs Normal
volcanoPlot(
coefs = lmTest$modelStats$disease_typecancer_coef,
p_vals = lmTest$modelStats$disease_typecancer_pval_FDR,
target_labels = lmTest$modelStats$target,
title = "Cancer vs Normal"
)
4.5.4 Saving Volcano Plots
# Automatically saves to file
volcanoPlot(
coefs = lmTest$modelStats$disease_typeinflam_coef,
p_vals = lmTest$modelStats$disease_typeinflam_pval_FDR,
target_labels = lmTest$modelStats$target,
title = "Cancer vs Normal",
plot_name = "figures/FDR_volcano_plot_inflam_vs_normal.pdf", # Filename (PDF, PNG, JPG, SVG)
data_dir = "figures", # Where to save
plot_width = 5, # Width in inches
plot_height = 5 # Height in inches
)
volcanoPlot(
coefs = lmTest$modelStats$disease_typecancer_coef,
p_vals = lmTest$modelStats$disease_typecancer_pval_FDR,
target_labels = lmTest$modelStats$target,
title = "Cancer vs Normal",
plot_name = "figures/FDR_volcano_plot_cancer_vs_normal.pdf", # Filename (PDF, PNG, JPG, SVG)
data_dir = "figures", # Where to save
plot_width = 5, # Width in inches
plot_height = 5 # Height in inches
)4.6 Visualization of Significant Targets
Visualize the proteins that show significant differences between Inflammation and Normal samples.
4.6.1 Inflammation vs Normal
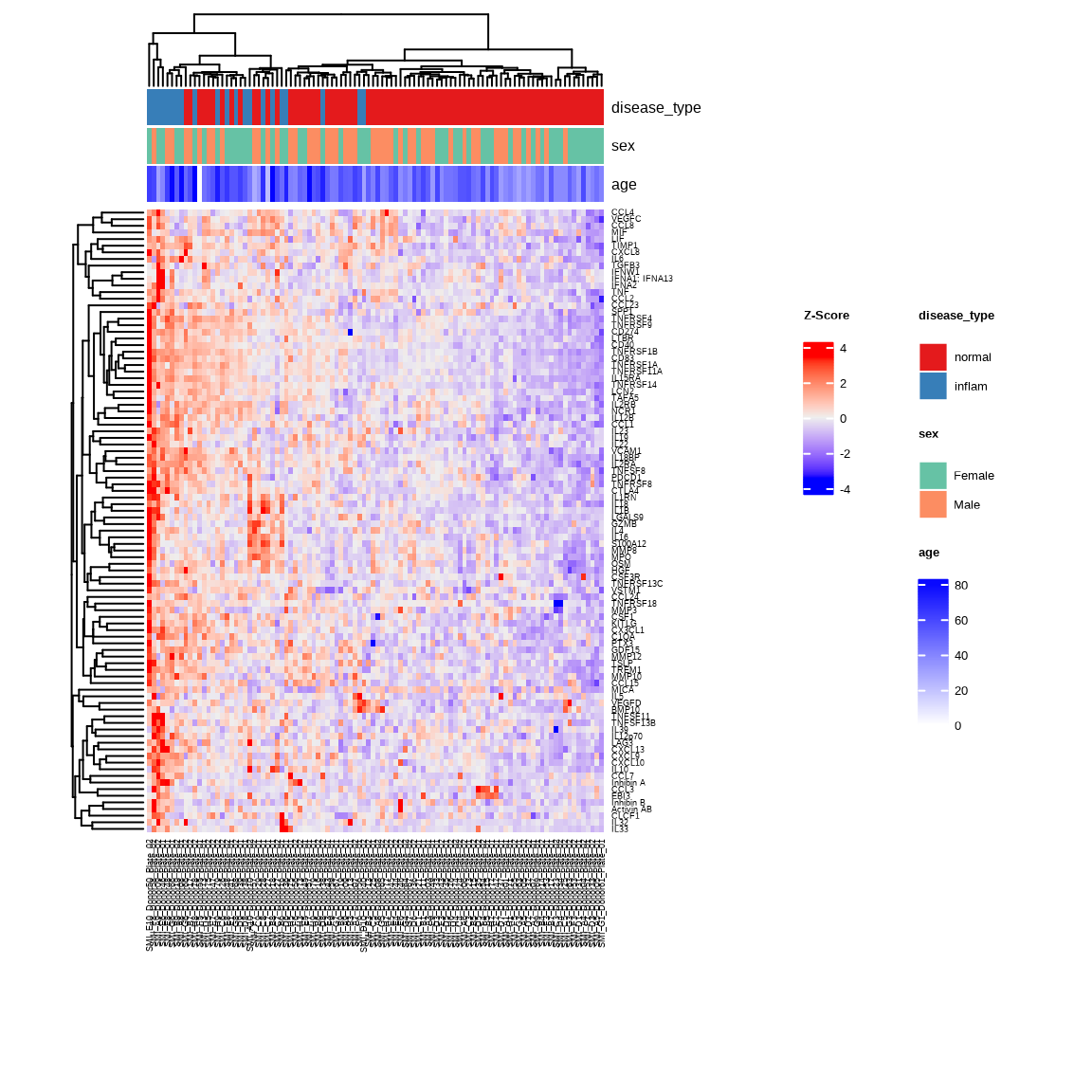
Heatmap
The heatmap shows expression patterns of upregulated proteins across Inflammation and Normal samples. Samples with similar expression profiles cluster together with unsupervised clustering.
inflam_samples <- metadata %>%
filter(disease_type %in% c("normal", "inflam")) %>%
pull(SampleName)
heatmap_inflam <- generate_heatmap(
data = data$merged$Data_NPQ,
sampleInfo = metadata,
sampleName_var = "SampleName",
sample_subset = inflam_samples,
target_subset = upregulated$target,
row_fontsize = 7,
col_fontsize = 7,
annotate_sample_by = c("disease_type", "sex", "age")
)
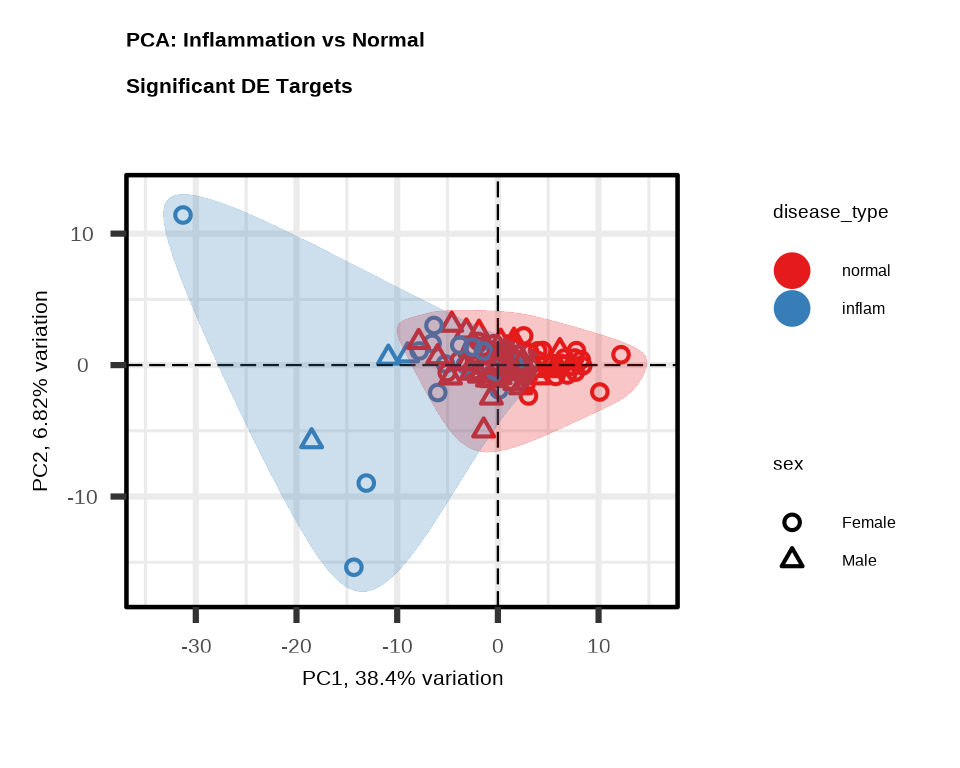
PCA
PCA shows how well the significant proteins separate Inflammation from Normal samples. Each point represents one sample.
pca_inflam <- generate_pca(
data = data$merged$Data_NPQ,
plot_title = "PCA: Inflammation vs Normal \nSignificant DE Targets",
sampleInfo = metadata,
sampleName_var = "SampleName",
sample_subset = inflam_samples,
target_subset = upregulated$target,
annotate_sample_by = "disease_type",
shape_by = "sex"
)
Boxplot: Upregulated Targets
Boxplots show the expression distribution of each upregulated protein in Inflammation versus Normal samples. Higher mean NPQ in Inflammation samples confirm upregulation.
data_long %>%
filter(Target %in% upregulated$target,
SampleName %in% inflam_samples) %>%
ggplot(aes(x = disease_type, y = NPQ, fill = disease_type)) +
geom_boxplot() +
facet_wrap(~ Target, scales = "free_y") +
labs(title = "Upregulated Significant Differential Expression Targets - Inflammation vs Normal",
subtitle = "with FDR-adjusted p < 0.05",
x = "Disease Type", y = "NPQ", fill = "Disease Type") +
theme(
plot.title = element_text(size = 16, face = "bold"),
plot.subtitle = element_text(size = 13),
axis.title.x = element_text(size = 14),
axis.title.y = element_text(size = 14),
axis.text.x = element_text(size = 12, angle = 45, hjust = 1),
axis.text.y = element_text(size = 12),
strip.text = element_text(size = 11),
legend.title = element_text(size = 13),
legend.text = element_text(size = 12)
)
4.7 Multiple Comparisons
When you have multiple disease types, you get results for each comparison:
# All comparisons vs "normal" (reference level)
comparisons <- c("inflam", "cancer", "kidney", "metab", "neuro")
for (comp in comparisons) {
coef_col <- paste0("disease_type", comp, "_coef")
pval_col <- paste0("disease_type", comp, "_pval_FDR")
if (coef_col %in% colnames(lmTest$modelStats)) {
n_sig <- sum(lmTest$modelStats[[pval_col]] < 0.05, na.rm = TRUE)
cat(comp, "vs normal:", n_sig, "significant proteins\n")
}
}#> inflam vs normal: 106 significant proteins
#> cancer vs normal: 69 significant proteins
#> kidney vs normal: 151 significant proteins
#> metab vs normal: 1 significant proteins
#> neuro vs normal: 0 significant proteins4.8 Model Considerations
4.8.2 Including Covariates
Always adjust for relevant covariates:
# Include covariates that might confound results
lmTest <- lmNULISAseq(
data = data$merged$Data_NPQ[, sample_list],
sampleInfo = metadata %>% filter(SampleName %in% sample_list),
sampleName_var = "SampleName",
modelFormula = "disease_type + sex + age + batch" # Added batch
)Common covariates:
- Technical: Batch, plate, run date
- Biological: Age, sex, BMI
- Clinical: Disease stage, treatment status
4.9 Interpreting Results
4.9.1 Effect Size (Coefficient)
- Positive coefficient: Protein is higher in test group vs reference
- Negative coefficient: Protein is lower in test group vs reference
- Magnitude: Larger value = bigger difference between groups
4.10 Exporting Results
4.10.2 Create Summary Table
# Count significant proteins per comparison
summary_table <- data.frame(
Comparison = character(),
Significant_Proteins = integer(),
Upregulated = integer(),
Downregulated = integer()
)
for (comp in comparisons) {
coef_col <- paste0("disease_type", comp, "_coef")
pval_col <- paste0("disease_type", comp, "_pval_FDR")
if (coef_col %in% colnames(lmTest$modelStats)) {
sig <- lmTest$modelStats %>%
filter(.data[[pval_col]] < 0.05)
summary_table <- rbind(summary_table, data.frame(
Comparison = paste(comp, "vs normal"),
Significant_Proteins = nrow(sig),
Upregulated = sum(sig[[coef_col]] > 0),
Downregulated = sum(sig[[coef_col]] < 0)
))
}
}| Comparison | Significant_Proteins | Upregulated | Downregulated |
|---|---|---|---|
| inflam vs normal | 106 | 106 | 0 |
| cancer vs normal | 69 | 68 | 1 |
| kidney vs normal | 151 | 151 | 0 |
| metab vs normal | 1 | 1 | 0 |
| neuro vs normal | 0 | 0 | 0 |
4.11 Complete Differential Expression Workflow
# Load libraries
library(NULISAseqR)
library(tidyverse)
# 1. Run differential expression
lmTest <- lmNULISAseq(
data = data$merged$Data_NPQ[, sample_list],
sampleInfo = metadata %>% filter(SampleName %in% sample_list),
sampleName_var = "SampleName",
modelFormula = "disease_type + sex + age"
)
# 2. Filter significant results
sig_inflam <- lmTest$modelStats %>%
filter(
disease_typeinflam_pval_FDR < 0.05,
abs(disease_typeinflam_coef) > 0.5
) %>%
arrange(disease_typeinflam_pval_FDR)
sig_cancer <- lmTest$modelStats %>%
filter(
disease_typecancer_pval_FDR < 0.05,
abs(disease_typecancer_coef) > 0.5
) %>%
arrange(disease_typecancer_pval_FDR)
# 3. Create volcano plots and save as PDF
volcanoPlot(
coefs = lmTest$modelStats$disease_typeinflam_coef,
p_vals = lmTest$modelStats$disease_typeinflam_pval_FDR,
target_labels = lmTest$modelStats$target,
title = "Cancer vs Normal",
plot_name = "FDR_volcano_plot_inflam_vs_normal.pdf",
data_dir = "figures",
plot_width = 5,
plot_height = 5
)
volcanoPlot(
coefs = lmTest$modelStats$disease_typecancer_coef,
p_vals = lmTest$modelStats$disease_typecancer_pval_FDR,
target_labels = lmTest$modelStats$target,
title = "Cancer vs Normal",
plot_name = "FDR_volcano_plot_cancer_vs_normal.pdf",
data_dir = "figures",
plot_width = 5,
plot_height = 5
)
# 4. Create heatmap and PCA of significant targets and save as PDF
## Get inflammation and normal sample name
inflam_samples <- metadata %>%
filter(disease_type %in% c("normal", "inflam")) %>%
pull(SampleName)
## Generate heatmap with annotations
heatmap_inflam <- generate_heatmap(
data = data$merged$Data_NPQ,
sampleInfo = metadata,
sampleName_var = "SampleName",
sample_subset = inflam_samples,
target_subset = upregulated$target,
annotate_sample_by = c("disease_type", "sex", "age"),
output_dir = "figures",
plot_name = "FDR_heatmap_inflam_vs_normal.pdf",
plot_width = 8,
plot_height = 6
)
## Generate PCA plot
pca_inflam <- generate_pca(
data = data$merged$Data_NPQ,
plot_title = "PCA: Inflammation vs Normal \nSignificant DE Targets",
sampleInfo = metadata,
sampleName_var = "SampleName",
sample_subset = inflam_samples,
target_subset = upregulated$target,
annotate_sample_by = "disease_type",
shape_by = "sex",
output_dir = "figures",
plot_name = "FDR_pca_plot_inflam_vs_normal.pdf",
plot_width = 5,
plot_height = 4
)
# 5. Create boxplot of significant targets and save as PDF
boxplot <- data_long %>%
filter(Target %in% upregulated$target,
SampleName %in% inflam_samples) %>%
ggplot(aes(x = disease_type, y = NPQ, fill = disease_type)) +
geom_boxplot() +
facet_wrap(~ Target, scales = "free_y") +
labs(title = "Upregulated Significant Differential Expression Targets - Inflammation vs Normal",
subtitle = "with FDR-adjusted p < 0.05",
x = "Disease Type", y = "NPQ", legend = "Disease Type") +
theme(strip.text = element_text(size = 8.5),
axis.text.x = element_text(angle = 45, hjust = 1))
print(boxplot)
ggsave(filename = "FDR_boxplot_inflam_vs_normal.pdf", plot = boxplot, device = "pef", path = "figures", width = 12, height = 9)
# 6. Export results
write_csv(lmTest$modelStats, "results/all_de_results.csv")
write_csv(sig_inflam, "results/sig_inflam_proteins.csv")
write_csv(sig_cancer, "results/sig_cancer_proteins.csv")
# 7. Print summary
cat("\nDifferential Expression Summary:\n")
cat("Inflammation vs Normal:", nrow(sig_inflam), "significant proteins\n")
cat("Cancer vs Normal:", nrow(sig_cancer), "significant proteins\n")4.12 Best Practices
Model Design
- Include relevant covariates to adjust for confounding
- Use appropriate reference levels
- Consider batch effects
Interpretation
- Using FDR-adjusted p-values for significance, accounting for multiple testing
- Consider both statistical significance AND effect size
- Validate key findings with independent methods
Reporting
- Document model formula used
- Report both FDR thresholds and fold-change cutoffs
- Include sample sizes per group
4.13 Common Issues
Problem: No significant proteins
- Check if groups are truly different (PCA)
- Sample size might be too small
- Effect sizes might be small
- Try less stringent FDR threshold or with unadjusted p values (exploratory only)
Problem: Everything is significant
- Check for batch effects
- Verify model is appropriate
- Consider more stringent thresholds
Problem: Results don’t match biology
- Check factor levels and reference group
- Verify metadata is correct
- Consider additional covariates
Continue to: Chapter 5: Longitudinal Analysis